PRIONES: PROTEÍNAS QUE COMEN CEREBROS
David Ismael Sánchez Mendoza

Una alarmante situación estalló en Gran Bretaña en 1986. Las vacas de este lugar empezaron a actuar fuera de lo común. Parecían totalmente ajenas a su ambiente y tenían problemas para moverse. Poco después fallecían.
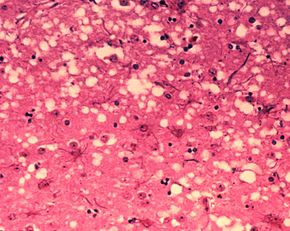
Al momento de examinar los sesos de estos bovinos, siempre presentaban la misma condición. Cavernas (huecos) que hacían parecer al cerebro como esponjas marinas. Por ello, los expertos decidieron nombrar a la patología «encefalopatía espongiforme bovina» o BSE (por sus siglas en inglés). Aunque para el público fue denominada «la enfermedad de las vacas locas».

Sin todavía haber hecho los estudios pertinentes, muchos científicos apuntaban o a un virus o un parásito, en este último caso capaz de ingerir células nerviosas. Sin embargo, tras los estudios moleculares no había ningún material genético ajeno al de las vacas.
Los investigadores que llevaron a cabo el experimento fueron rápidamente desacreditados por haber procesado las muestras de manera errónea, así que otros científicos tomaron la batuta de repetir la prueba. Obtuvieron el mismo resultado.
Sin respuestas y ante el aumento del brote epidémico, se estima que para 1993 Gran Bretaña tuvo que sacrificar más de un millón de cabezas de ganado a causa de la infame patología, sin contar las que ya habían sido procesadas y exportadas con la enfermedad al resto de Europa.
En este punto volveremos atrás en el tiempo, específicamente al año 1982. En ese entonces, el neurólogo estadounidense Stanley Pruiser y su equipo publicaron un trabajo en el cual afirmaban que una proteína con capacidad infecciosa había sido aislada y purificada (ya que había infectado a un hámster). Esta proteína infecciosa ocasionaba una enfermedad neurodegenerativa llamada scrapie (visna) en ovejas y cabras. Él decidió llamar a dicha proteína «prión», por proteinaceous infectious particle.
No obstante, la comunidad científica estaba muy escéptica respecto a la capacidad de las proteínas de infectar. Sin más, sus resultados fueron ignorados.
O al menos durante mucho tiempo. En abril de 1996, la revista británica Lancet, a modo de héroe incógnito, realizó un recuento de tres investigaciones que narraban patologías neurodegenerativas que parecían transmisibles. Todo esto ya que se había descrito que 10 individuos de Gran Bretaña y Francia habían fallecido tras previos cuadros de demencia y pérdida locomotora. Las autopsias revelaron que todos tenían las perforaciones cefálicas anteriormente identificadas en las vacas, Como resultado, se atribuyeron las muertes al consumo de carne contaminada proveniente de las vacas locas.
La primera de las menciones hechas por Lancet databa de 1920, cuando el neurólogo alemán Hans Creutzfeldt y un poco después Alfons Jakob, documentaron estas perforaciones encefálicas por primera vez. Como dato curioso se le nombraría «enfermedad de Creutzfeldt-Jakob» o CJD (por sus siglas en inglés) a esta patología en el humano en honor a ellos.
La segunda corresponde al médico Carlenton Gajdusek, el cual en 1960 fue a Papúa Nueva Guinea para estudiar el «Kuru», un padecimiento que afectaba a la población nativa. El demostró que éste era una enfermedad neurodegenerativa que se transmitía durante el ritual funerario que las personas practicaban. En dicho ritual se comían el tejido cerebral del cadáver a modo de «heredar su sabiduría».

Gajdusek además comprobó en 1968 que al introducir extractos de biopsia cerebral con Kuru en un animal, éste desarrollaba la neurodegeneración con sus respectivas cavernas.
La última por supuesto fue la de Pruiser, quien tras resolverse el enigma recibió el Premio Nobel en Fisiología o Medicina en 1997, al siguiente año de la reseña.
Así, con todas las piezas científicas e investigativas en sus lugares, se logró saber lo que había ocurrido con las vacas. Todo fue culpa de las enfermedades priónicas.
Una de las peculiaridades de éstas enfermedades es que, a pesar de que todas generan neurodegeneración, reciben nombres diferentes según la especie que afecta:
- Scrapie – ovejas y cabras
- Encefalopatía Espongiforme Bovina – bovinos
- Enfermedad de Creutzfeldt-Jakob – humanos
- Caquexia (o Desgaste) crónica – venado y alce
- Encefalopatía transmisible del Visón – visón
- Encefalopatía espongiforme felina – gatos
¿Cómo empezó todo?
Se descubrió que las vacas habían sido alimentadas con restos de oveja que habían padecido scrapie. Tras consumir la médula espinal se infectaron con el prión y éste les ocasionó encefalopatía espongiforme bovina.
A su vez, el consumo de sesos de las vacas infectadas por parte de esas 10 personas (así como la práctica del Kuru transmitió el prión a esos humanos y les originó la enfermedad de Creutzfeldt-Jakob variante o «CJD variante» el término variante hace alusión a que por fue causada por restos contaminados.
Vale aclarar que el humano por sí mismo (sin consumir alimentos contaminados) puede originar un prión y por ende una enfermedad priónica que recibe el nombre de enfermedad de Creutzfeldt-Jakob clásica o «CJD clásica».
¿Qué es un prión?
En pocas palabras es una proteína cuya conformación tridimensional está alterada, es decir, no es la que debería poseer.
Hablando explícitamente de los humanos, ésta posibilidad se encuentra en el cromosoma 21, en un gen denominado PRNP, el cual se expresa en el tejido cerebral.
Dicho gen normalmente produce una sialoglucoproteína designada PrPC (prion protein celular), que reside en la superficie de las neuronas y cuya función todavía es desconocida (algunos suponen que está vinculada al potencial de transmisión)
Imaginemos que durante el plegamiento de PrPC alguna condición dentro de la célula evita que ésta tome su configuración tridimensional correcta. El resultado será entonces PrPSC (prion protein scrapie), el prión responsable del CJD clásico.
Si bien ambas tienen la misma secuencia de aminoácidos, sus estructuras tridimensionales serán muy distintas. Mientras PrPC tiene forma de cuerda enrollada sobre sí misma, PrPSC dará la apariencia de láminas plegadas una detrás de otra. Estas distinciones también cambiarán aspectos físicos y de integración entre ellas.
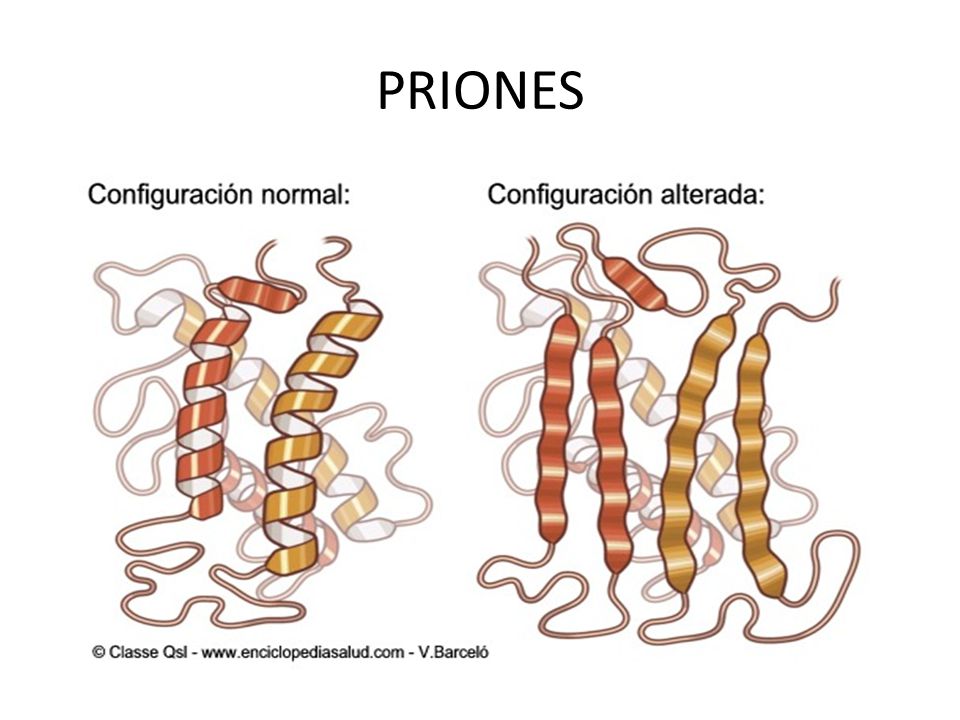
PrPc (o la proteína no priónica) siempre se encontrará sola, sin estar unida a ninguna otra compañera PrPC (monómero) y será fácilmente digerible por proteasas. Diferente será el caso de PrPsc (la proteína priónica), la cual se unirá a otras iguales a ella y formará fibrillas insolubles resistentes a la ruptura.
Es aquí donde entramos en el terreno del «agente infeccioso», ya que las fibrillas PrPsc pueden «infectar» a las PrPc mediante la alteración de su configuración, convirtiendo así la versión no priónica en la priónica, a modo de reacción en cadena.
Y son estas fibrillas las causantes de las cavernas en los cerebros de los afectados, ya que el aumento en número de éstas ocasiona la muerte celular de la neurona. Para colmo, los priones conocidos hasta la fecha, no son inmunógenos, es decir, no generan una respuesta inmunitaria y por ende el cuerpo no los rechaza.
De esta forma, la ciencia dio lugar a un descubrimiento histórico e impensado en su momento, pero que en la actualidad forma parte de ese gran conglomerado de conocimiento sobre biología celular y microbiología.
A pesar de esta penosa situación y el gran descubrimiento que supuso, no hay nada que temer. Si bien, la CJD clásica es una enfermedad letal, a nivel estadístico es muy infrecuente. Por ejemplo, en Estados Unidos y Europa, si hablamos de un individuo que supera los 50 años, ésta se presenta en una por cada millón de habitantes. En cambio, si la edad es igual o menor a 50, la incidencia será de 1 por cada doscientos millones de habitantes. Hablando de la CJD variante, los productos alimentarios de animales contaminados pueden transmitir la enfermedad, pero con los tratamientos clásicos que se le dan a dichos productos (como la pasteurización) el prión queda desactivado.
El conocimiento nos ayuda a vivir más tranquilos, sin preocuparnos más por las proteínas que comen cerebros.
Referencias.
- Karp, G. (2010). Biología Celular y Molecular. Conceptos y Experimentos. Mc Graw Hill. 6ta edición. México.
- Cooper G, Hausman R. (2011). La Célula. Marbán. 5ta edición. España.
- Brooks G, Carrol K, Butel J, Morse S, Mietzner T. Jawetz, Melnick y Aldelberg Microbiología Médica. (2011). Mc Graw Hill. 25va edición. México.
- Lodish H, Berl A, Matsudaira P, Kaiser C, Krieger, Scott M, et al. (2011). Biología Celular y Molecular. Editorial Médica Panamericana. 5ta ed. Buenos Aires, Argentina.
- Zabel M & Reid C. (2015) A brief history of prions.[En línea]. Pathogens and Disease. Oxford, Inglaterra, 73 (9): ftv087. doi:10.1093/femspd/ftv087
- Terry, C, & Wadsworth, J. ( 2019). Recent Advances in Understanding Mammalian Prion Structure: A Mini Review. Frontiers in molecular neuroscience. [En línea]. Frontiers in Molecular Neuroscience. Suiza, 12 (1): 169. doi:10.3389/fnmol.2019.00169
 David Ismael Sánchez Mendoza es licenciado en Citotecnología (Universidad Arturo Michelena, Venezuela) con diplomado en Biología Molecular y Análisis Clínicos. Bloguero científico desde 2016 con Ciencia de El Principito a El sueño y el vuelo. También divulga ciencia en su cuenta de Instagram: @soyciencia.
David Ismael Sánchez Mendoza es licenciado en Citotecnología (Universidad Arturo Michelena, Venezuela) con diplomado en Biología Molecular y Análisis Clínicos. Bloguero científico desde 2016 con Ciencia de El Principito a El sueño y el vuelo. También divulga ciencia en su cuenta de Instagram: @soyciencia.
AGRADECIMIENTOS
La Fundación Persea agradece la infinita generosidad de sus patrocinadores: Carlos Ortega Sr., Sobella Mejías, Solmar Valera, Jiří Svozilík, Leonardo Quevedo, Héctor Pittman Villarreal, My fit body project y Vicente Di Clemente.